As part of my blog, I’d like to share with you, my readers,
what I’m actually doing now that I’m settled into the institute. For the sake
of my research, I won’t be discussing any results. That’s for once the seminars
begin. For the sake of your sanity, I’m also keeping the methods very
general, since I want this to reach to everyone.
In plant pathology, it is said that the journey of a
thousand experiments begins with a single spore. Ok, no one in my field says
that, I just needed a cheesy introduction.
The first step when I arrived to JAAS was to take the collection of
isolates and culture them from a single spore. This technique is appropriately called
the single spore isolation. This is done with most fungi for various reasons.
The most important is uniformity. If you have a pure culture (that is, only one organism
growing in your plate), there is still no way to ensure that the one organism is a
single lineage or if it’s a diverse population. You could have 15 different
individuals of the same organism in there, and that could cause some
inconsistencies with experiments. Sometimes the fungi is grey or white, next
time it’s green!
 |
| This is NOT a pure culture. Some contaminants are clearly present |
 |
| This is a pure culture. There's only one organism growing in there. But is it all the same organism or a population of the same organism? |
First step: Get spores
It may surprise you that in order to do a single spore
isolation, you need spores. Now, that can be challenging for some fungi, since
they don’t make spores. I had a labmate who worked on a pathogen called Sclerotium rolfsii that does not produce
spores. There are some methods that can be used if that happens. However,
that’s a whole different topic that I’m glad to avoid.
For my fungus, it produces two types of spores, asexual
(called conidia) and sexual (called ascospores). Each kind of these spores is
produced in a separate structure. Conidia are borne on a structure called a
pycnidia, whereas ascospores are borne on either a perithecia or a pseudothecia
(don’t get too hung up on the names, it has to do with the shape and structure
of the fruiting body). For the sake of simplicity, conidia are the ones I want.
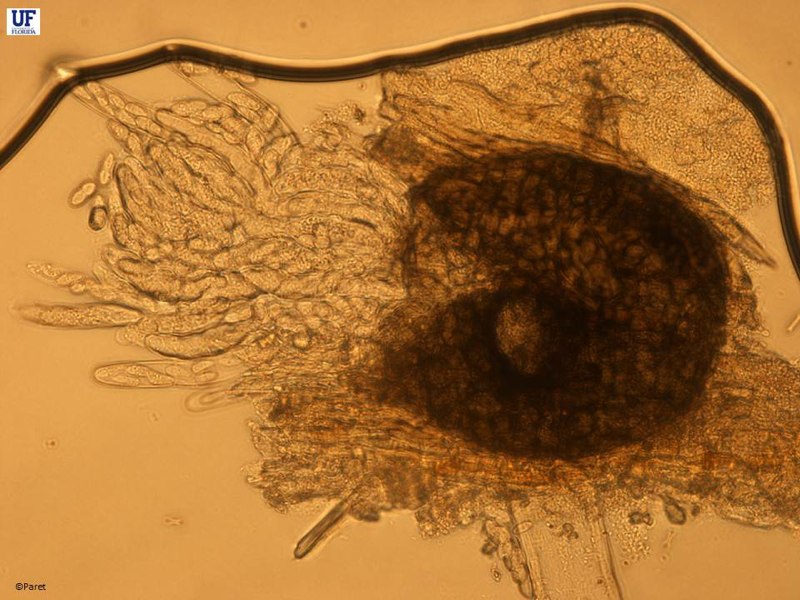 |
| This a perithecium with ascospores emerging. |
 |
| Pycnidia with conidia being released. Each spore is a clonal copy of its parent |
So how do I get these conidia?
The fungus has to be producing these pycnidia in order to
get spores. There are a few methods that I use to get said spores. First,
adding light to the growth chamber encourages the fungus to grow. Just as
plants respond to light, so too will fungi. It isn’t essential for their
growth, but it triggers certain responses, including pycnidiation. Adding in UV light also encourages
pycnidia to form. Another method is to grow the fungus on a minimal media. If
the fungus runs out of food, the next step is to make spores to find a better
environment.
I have spores! So now what?
So once we have spores, we have to count them. The best way is to take some of the spores from a petri plate and scrape them into some sterile water. At high
enough concentrations, you can actually measure optical density (i.e. how
“blurry” the spores are in water). However, there’s another way if fungus
doesn’t produce billions of spores. There is a device called a hemocytometer (sometimes spelled with an "a" hemacytometer).
Originally, it was produced to count red blood cells. It is also used to do
sperm counts in both animals and humans. Fellas, if you ever go in for
fertility testing, there’s a good chance that one of these will be used to assess your sperm.
 |
| Thanks Wikipedia! |
So, the spore suspension is placed on the hemocytometer, and
on the surface, there is a grid pattern. That grid pattern is viewed under a
compound microscope. Within each grid is a known volume, typically 0.1µl. That’s one
microliter, take your liter of cola and divide it by one million, Farva. We count the
number of spores and from that concentration, we can adjust out suspension of
spores to a desired amount. I’ll spare you all the math.
 |
| A typical grid pattern on a hemacytometer, The red area (letter A for color blind) is 0.1ul in volume |
I did the math! Now what do I do?
Once the spore suspension has been adjusted the suspension
is then dropped onto another petri plate. For the sake of simplicity, I’ll go
through an example. Say the suspension is adjusted to 1000 spores per 1ml of
water, each one of those little droplets is going to be 1µl, which is 1/1000 of
1ml. In theory, there should be 1 spore within that aliquot. This
is repeated several times as there is a lot of variance within that suspension.
The conidia often come out of the pycnidia in a ribbon and are held together by a
gel-like substance; adding a detergent reduces the gelling. Often, it’s called a “spore horn”. Also, this assay does not distinguish between viable cells and dead ones, so more chances are better chances. After the suspension has
been dropped, the plate is left alone for 24 hours.
 |
| If you mess with the spores, you sometimes get the horns. |
I waited 24 hours, what’s the next step?
After the 24 hours, the plate is observed under the
microscope and the spores will have germinated. Now comes the tricky part, an
aliquot containing exactly one spore must be found. Given the variance, it’s
not uncommon to find one aliquot with 6 germinated spores and the next aliquot
to have none. After a bit of patience and luck, one perfect aliquot is found! We use a
cork borer to carefully cut the agar chunk out of the original plate and that chunk is
then plated onto a fresh, clean plate. After 48 hours, the single spore will
have grown to the size where it is detectable to the naked eye.
I successfully single-spored, now what do I do with it?
Now you can run all of your experiments!
This is only the first step in the research journey. From
start to finish it takes about 3 days to complete, assuming the culture is already creating spores. For the sake of
consistency, nothing can be done before the single spore isolation, so it
becomes a crucial task if working with fungi. I plan on generally describing
some of the other experiments I’m running in the lab. I know I used a lot of
technical terms, so feel free to ask for clarification in the comments.
Mason
No comments:
Post a Comment